3 Ways To Improve Flow Cytometry Troubleshooting

Flow cytometry troubleshooting is a regular part of my job. I spend a lot of time working with my colleagues and clients retrospectively troubleshooting their data. This involves trying to understand and explain what might’ve happened during the data acquisition that led to the results in question.
During these discussions, I give them tips and strategies to improve their acquisition and data quality over time because we want to make sure that people have the highest quality data they can. This means that the researchers should understand and follow the best practices and are taking the time to make sure that their data is collected correctly.
There are three main areas that you need to think about when troubleshooting your experiments…
3 Keys To Flow Cytometry Troubleshooting
1. What do you before you start collecting data?
When you sit down at the cytometer, what are the steps that you go through to make sure the cytometer is ready?
Do you check the quality control logs?
Do you ask the operators for the quality control to see how the machine’s behaving since the last time you ran the system?
Or, better yet, do you have some sort of quality control built into your assay?
This could include a bead, such as the Spherotech peak 6 bead, which is a very useful tool. Any standard bead you choose is useful for QC, as long as you use it in a consistent manner.
Here are the steps I recommend you follow
- When you sit down at the instrument put on a new tube of distilled water. While you are setting up your templates, let this run on high to make sure that whoever ran the system last cleaned it properly, and there’s no bleach there.
- Put your QC bead (peak 6 bead) on and open your target template for the assay. Run that bead to make sure that you’re hitting the target values that you set when you established the baseline during the development phase of your panel.
- Monitor what voltages that you’re using and if they’ve been significantly different than the previous runs, stop there before you do anything else. You need to figure out what happened.
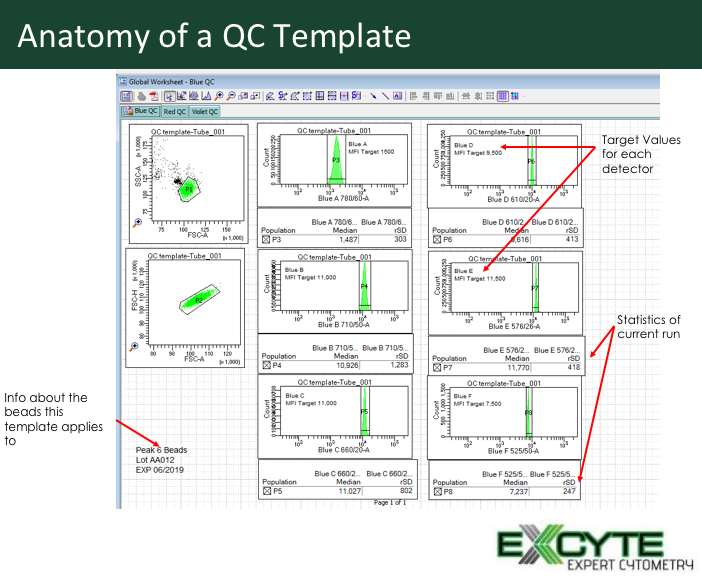
Figure 1: Anatomy of a User-defined QC template
Was there an issue with the laser? Was there an issue with the PMT? Was the system not clean? Is there something going on that I need to address before I go on to the second point?
Figure out the problem before starting your experiment to improve your flow cytometry troubleshooting process.
2. Ensure you have plots of time vs fluorescence for each of the lasers you are using.
The first thing you really want to do is make sure you have plots of time versus fluorescence for each of the lasers that you’re looking at and that you are monitoring that fluorescence to make sure you’re not seeing any problems.
Two big problems you’re going to worry about are clogs in front of the flow cell and clogs in the back end of the flow cell.
Typically, a clog in the front end of the flow cell is going to manifest itself by you not seeing any events at all.
If that happens you have to take your tube off and do some cleaning procedures. This could be priming to flush the SIP or using any sort of automatic declogging protocol that the system may have.
If it gets really bad, you might have to use something like Contrad or bleach to clean out even more, and eventually, you may have to escalate it up to your core staff.
Now if you have a block on the far side of the flow, this causes a buildup of back pressure because the clog has narrowed the tube. The backpressure reducing the speed of the flow cytometer, resulting in the cells missing their time delay windows.
This typically will manifest itself by you losing signal at the farthest laser from the target laser, typically the blue laser is your target laser.

Figure 2: Good vs bad flow. The plot on the left shows good, uniform flow while on the right are the consequences of back pressure slowing the flow rate down, making the cells miss the time delay window.
So say on your green laser, that’s three lasers out, you lose that signal first. You can see that happen when you’re watching your plots.
I also like to encourage people to run their samples in tubes. I know the HTS has an allure of being able to walk away, but if you walk away from your system, you cannot monitor what’s going on, and you can lose something.
Also, a lot of the HTS systems don’t agitate the samples efficiently, so the sample is not always suspended. If you’re running large volumes, the cells might start to settle and you’re going to lose those cells because some of those cells have settled in the dead volume.
So, as much as HTS can save time, it’s better to run the samples in the tube so you can see what’s going’ on.
3. Apply appropriate gating procedures.
What happens if you look at your data after you’ve acquired it? One of the first strategies is to do flow stability gating.
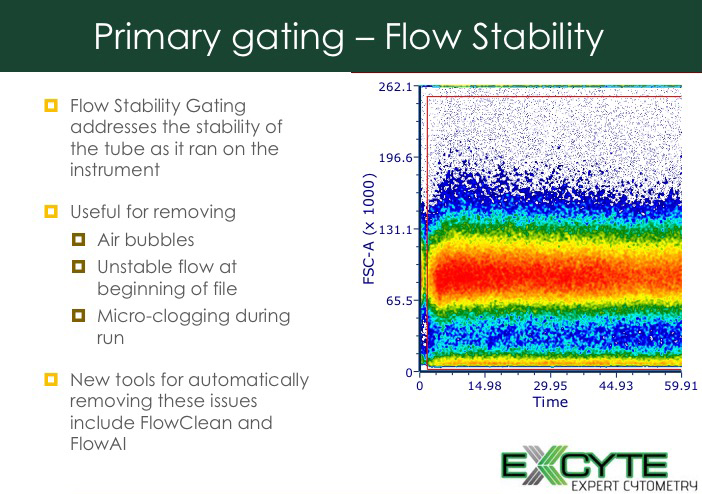
Figure 3: Flow stability gating
If you have walked away from monitoring your run, this flow stability gate is especially important. It lets you identify issues through the run and eliminate them.
Next, go through the rest of your gating strategies and make sure that all the parameters look good. This is another place where having a control, such as a reference control that you know how this control behaves becomes very useful.
Figure 4: Tracking reference control behavior.
With the reference control, when you initially run this tube, it will help identify any issues on the system. During analysis, it helps ensure that the expected gating profiles are being maintained and data within the acceptable range is being generated.
With flow cytometry troubleshooting, the focus is often on fluidics. If you sit down and think about your workflow, and how you might want to add a couple of little tweaks here and there which will ultimately help you improve the quality of your data as well as aid you in identifying issues before they become problems your troubleshooting will be much smoother. Consider these three things, what do you before you start collecting data, ensure you have appropriate plots of time vs fluorescence for each of the lasers your using and apply appropriate gating procedures.
Want to learn more about flow cytometry troubleshooting and get access to all of our advanced materials including 20 training videos, presentations, workbooks, and private group membership, get on the Flow Cytometry Mastery Class wait list.

ABOUT TIM BUSHNELL, PHD
Tim Bushnell holds a PhD in Biology from the Rensselaer Polytechnic Institute. He is a co-founder of—and didactic mind behind—ExCyte, the world’s leading flow cytometry training company, which organization boasts a veritable library of in-the-lab resources on sequencing, microscopy, and related topics in the life sciences.
More Written by Tim Bushnell, PhD











