How To Profile DNA And RNA Expression Using Next Generation Sequencing (Part-2)

In the first blog of this series, we explored the power of sequencing the genome at various levels. We also dealt with how the characterization of the RNA expression levels helps us to understand the changes at the genome level. These changes impact the downstream expression of the target genes. In this blog, we will explore how NGS sequencing can help us comprehend DNA modification that affect the expression pattern of the given genes (epigenetic profiling) as well as characterizing the DNA-protein interactions that allow for the identification of genes that may be regulated by a given protein.
DNA Methylation Profiling or Epigenetic Profiling
NGS can be adapted to profile DNA methylation either through an enrichment (using methyl CpG antibody or methyl-CpG-binding protein) or by bisulfite sequencing.

Figure 1: Different methods of NGS DNA Methylation profiling.
1. Bisulfite Sequencing
Bisulfite treatment of DNA converts unmethylated cytosines to uracil, while methylated cytosines remain the same. Uracil bases are then identified as thymine in the sequencing data, which could be used to identify the location and percentage of methylated cytosines. NGS-based bisulfite sequencing — whether whole-genome or targeted — makes it possible to profile genome-wide cytosine methylation at single-base resolution.
Types of Bisulfites sequencing:
a. Whole-Genome Bisulfite Sequencing (WGBS)
Currently, WGBS is the most comprehensive way to profile DNA methylation at base-pair resolution. However, the required depth (minimum 30x) makes it cost-prohibitive. Thus, other enrichment methods have been devised to reduce the cost of methylation profiling, especially when 100% coverage or base-pair resolution is not necessary.
b. Reduced Representation Bisulfite Sequencing (RRBS)
RRBS relies on restriction enzymes such as MspI (CCGG) or BglII (AGATCT), which tend to cut inside or near CpG islands and promoter regions regardless of methylation status. Subsequently, fragments between 40 – 220 bp are isolated and end-repaired, then treated with bisulfite and amplified with PCR. RRBS using MspI captures approximately 80% of CpG islands and 60% of promoter regions in human genomes.
2. Methylated DNA-enriched Sequencing
a. MethyCap-Seq
This sequencing uses the Methyl-CpG-binding (MBD) domain of MeCP2 to capture methylated DNA on magnetic beads. After the captured DNA is enriched with magnetic capture, the bound DNA is eluted with a high-salt solution and then used for NGS. While this is a cost-effective method, the current resolution is ~150 bp, so it is suitable for fast, large-scale, and low-resolution studies.
b. Methylated DNA Immunoprecipitation-Seq (MeDIP-Seq)
It uses an anti-methylcytosine antibody to immunoprecipitate DNA with methyl CpG. While MeDIP-Seq can be relatively inexpensive, it can yield resolutions of between 100 – 300 bp.
DNA-protein Interaction Profiling
Due to the quantitative nature of NGS, chromatin immunoprecipitation-enriched DNA can be sequenced with NGS to profile any genomic regions bound by the proteins of interest that can either be recognized with an antibody or tagged with an epitope. These include DNA-binding proteins, transcription factors, histones, histone variants, specific histone modifications, and nucleosomes.
1. ChIP-Seq (Chromatin Immunoprecipitation Sequencing)
To create a ChIP enriched library, DNA-bound proteins are cross-linked to DNA using formaldehyde, before the chromatin is cleaved. The sample is then enriched using immunoprecipitation with an antibody specific to the protein or protein modification of interest. Subsequently, the crosslinks are reversed, and then the ChIP enriched library can be assayed using quantitative PCR, microarray, or NGS.
Difference between ChIP-chip Vs. ChIP-Seq
ChIP-chip resolution is limited by the probes’ fragment sizes on the arrays, whereas ChIP-Seq can provide single-nucleotide resolution. ChIP-Seq requires much less input DNA and provides signals with an unlimited dynamic range, depending on the sequencing depth. Additionally, ChIP-Seq makes it possible to profile repetitive regions – these are often omitted from the microarrays. Repetitive regions that are often important for epigenetic control, such as heterochromatin or microsatellites, may only be mapped with NGS.
In addition to identifying genomic regions bound by the proteins, ChIP-Seq can provide insights into the functions of the DNA-bound proteins themselves. For example, ChIP-Seq data can be used to identify the cognate binding motifs of the DNA-binding proteins. This sequence data can also be used to globally infer distances between the binding sites and genomic features, such as transcription start sites, exon-intron boundaries, 3’end of genes, and from other known binding sites.
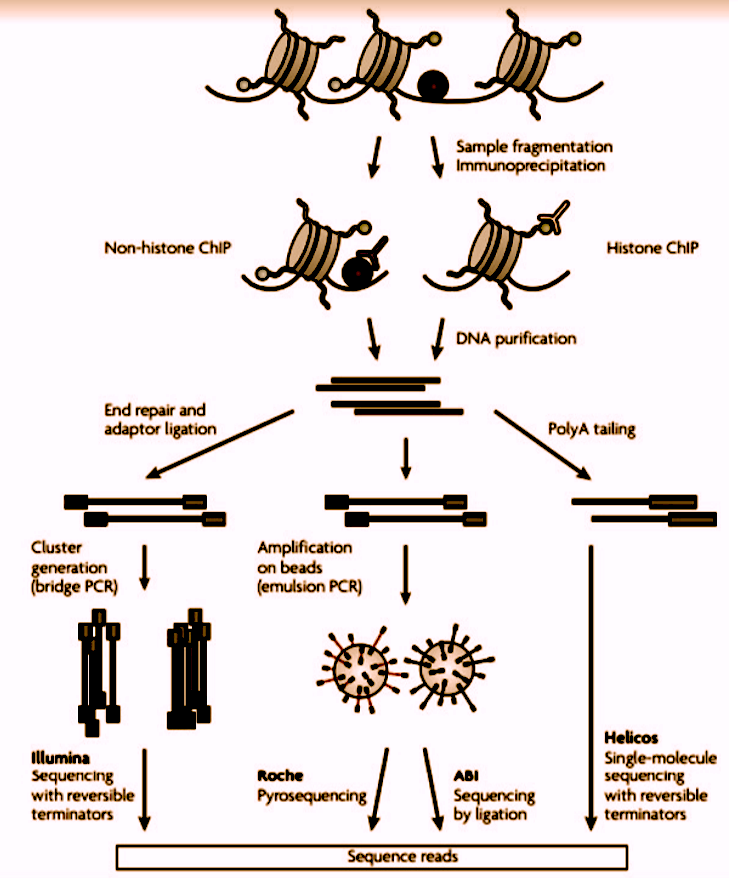
Figure 2: A representation of Chip sequencing
- Micrococcal Nuclease-Seq (MNase-Seq)
Nucleosome occupancy can tell us about regions of active genes and chromatin structure in eukaryotes. NGS allows us to profile the nucleosome occupancy by sequencing the micrococcal nuclease (MNase)-digested genomic DNA. MNase prefers to digest linker DNA between histone octamers unoccupied by other proteins.
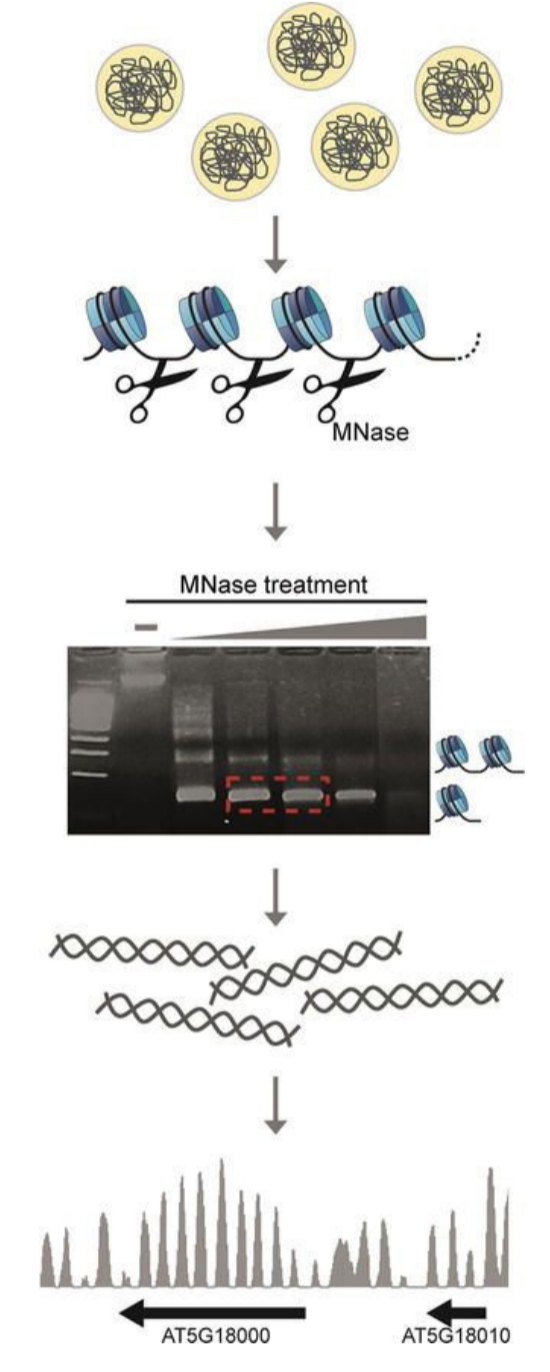
Figure 3: The workflow of an MNase protection assay
DNA is crosslinked to the protein using formaldehyde before MNase digestion. Once the digestion step is complete, the crosslinks are reversed. Then, the digested DNA is run on a gel to select the desired digested products, which are then purified and subsequently used for NGS. To control for MNase sequence bias, GC/AT preference, and other technical biases, it is necessary to concurrently sequence the genomic DNA from the same sample without crosslinking – and compare them during the analysis process.
Concluding Remarks
Over the course of these two blog posts, we have explored the power of NGS sequencing at several levels, from whole-genome sequencing, down to characterizing epigenetic differences that impact gene expression. NGS sequencing allows scientists to get a deeper holistic understanding of the genome, and variations that may be markers for the disease. No other technique can provide such a complete picture in a relatively short time frame. As costs continue to decrease, these techniques will continue to have a greater role in areas such as drug discovery, clinical diagnostics, and ultimately personalized medicine. Stay tuned to this blog for more information on these and many other techniques being developed in the world of NGS sequencing.
To learn more about gene prediction and how NGS can assist you, and to get access to all of our advanced materials including 20 training videos, presentations, workbooks, and private group membership, get on the Expert Sequencing wait list.

ABOUT DEEPAK KUMAR, PHD
GENOMICS SOFTWARE APPLICATION ENGINEER
Deepak Kumar is a Genomics Software Application Engineer (Bioinformatics) at Agilent Technologies. He is the founder of the Expert Sequencing Program (ExSeq) at Cheeky Scientist. The ExSeq program provides a holistic understanding of the Next Generation Sequencing (NGS) field - its intricate concepts, and insights on sequenced data computational analyses. He holds diverse professional experience in Bioinformatics and computational biology and is always keen on formulating computational solutions to biological problems.
More Written by Deepak Kumar, PhD











