When To Use (And Not Use) Flow Cytometry Isotype Controls

Antibodies can bind to cells in a specific manner – where the FAB portion of the antibody binds to a high-affinity specific target or the FC portion of the antibody binds to the FcR on the surface of some cells.
They can also bind to cells in a nonspecific manner, where the FAB portion binds to a low affinity, non-specific target. Further, as cells die, and the membrane integrity is compromised, antibodies can non-specifically bind to intracellular targets.
The question has always been how to identify and control for the nonspecific antibody binding observed.
This led to many research groups using a control called the Isotype control.
The concept of this control is that an antibody targeting a protein not on the surface of the target cells, has the same isotype (both heavy and light chain) as the antibody of interest. When used to label cells, those that showed binding to the isotype, would be excluded as they represented the non-specific binding of the cells.
When Isotype Controls Were Everything
Isotype controls were once THE negative control for flow cytometry experiments.
They are still very often included by some labs, almost abandoned by others, and a subject of confusion for many beginners. What are they, why and when do I need them? Are they of any use at all, or just a waste of money?
Most importantly, why do reviewers keep asking for them when they review papers containing flow data?
Isotype controls were classically meant to show what level of nonspecific binding you might have in your experiment. The idea is that there are several ways that an antibody might react in undesirable ways with the surface of the cell.
Not all of these can be directly addressed by this control (such as cross-reactivity to a similar epitope on a different antigen, or even to a different epitope on the same antigen). What it does do is give you an estimate of non-specific (non-epitope-driven) binding. This can be Fc mediated binding, or completely nonspecific “sticky” cell adhesion.
In order to be useful, the isotype control should ideally be the same isotype, both in terms of species, heavy chain (IgA, IgG, IgD, IgE, or IgM) and light chain (kappa or lambda) class, the same fluorochrome (PE, APC, etc.), and have the same F:P ratio. F:P is a measurement of how many fluorescent molecules are present on each antibody.
This, unfortunately, makes the manufacture of ideal isotype controls highly impractical.
There is even a case to be made that differences in the amino acid sequence of the variable regions of both the light and heavy chains might result in variable levels of undesirable adherence in isotypes versus your antibody of interest.
Moving Beyond Isotype Controls
For these reasons, many in the field are moving beyond the isotype control. With some suggesting they be left out of almost all experiments.
If you spend any time browsing the Purdue Cytometry list, you’ll see these same arguments presented in threads about isotype controls.
The following paper presents options for controls in several categories, the options available, and pros and cons of each option. The section on isotype controls summarizes the problems with the use of isotype controls very clearly…
Flow cytometry controls, instrument setup, and the determination of positivity.
Additionally, the following paper presents options for controls in several categories, the options available, and pros and cons of each option…
Considerations for the control of background fluorescence in clinical flow cytometry.
The section of the above paper focusing on isotype controls summarizes the problems with their use very clearly.
The article also illustrates difference in undesirable binding at different levels using the same clone from different manufacturers.
See the figure below for an example of how even the same isotype control clone can result in highly variable levels of undesirable staining.

If you do use isotype controls in your experiment, they must match as many of the following characteristics as possible for your specific antibody—species, isotype, fluorochrome, F:P ratio, and concentration.
9 Tips For Using (Or Not Using) Isotype Controls
1. You certainly don’t need them for things that are clearly bimodal. If you are looking for T cells and B cells in peripheral blood the negative cells also in the circulation provide gating confidence. As seen in the example below, it is extremely easy to pick out CD4 and CD8 positive cells in the sample of lysed mouse blood.
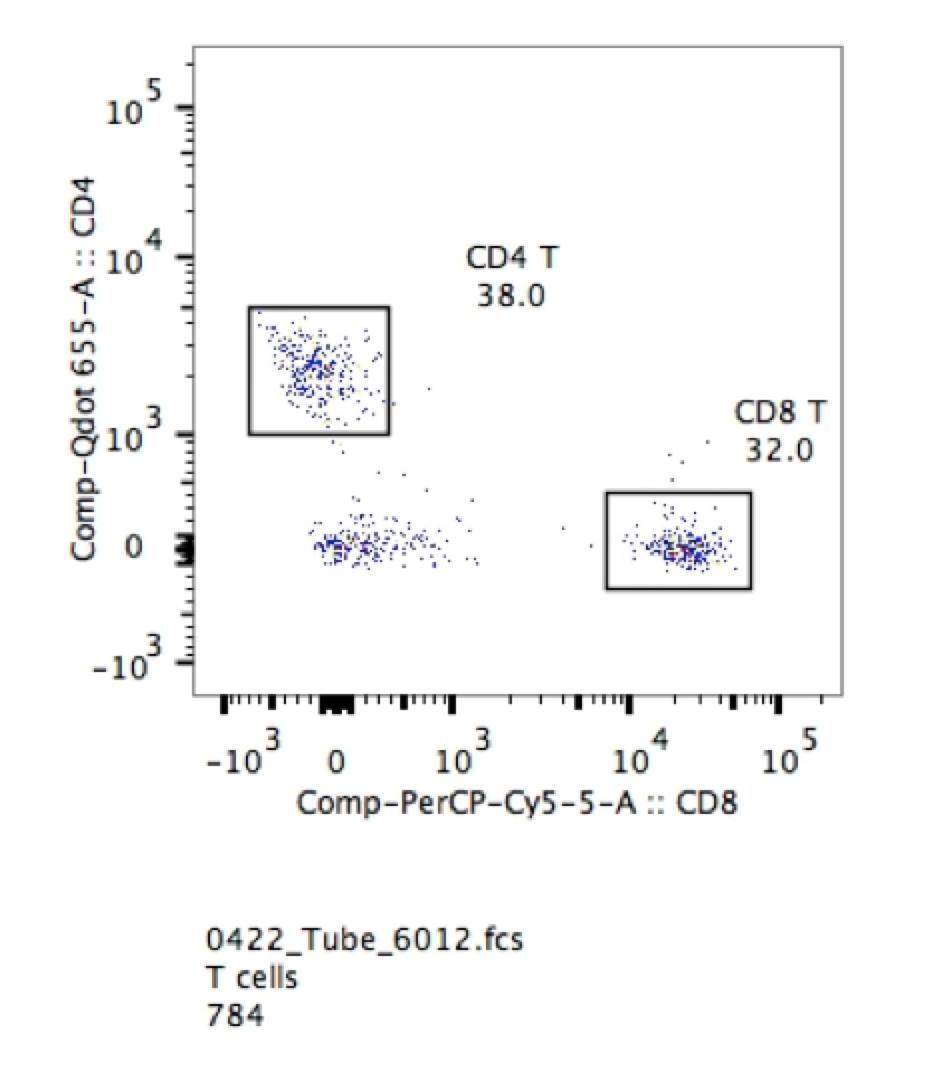
2. If you are using post-cultured cells, the isotype control might give you some information about the inherent “stickiness” of your cells. However, this is not meant as a value you can subtract from your specific antibody sample in terms of fluorescence intensity or percent positive. This is simply a qualitative measure of “stickiness” and the effectiveness of blocking in your protocol.
3. If you are using multiple dyes in your search, and your concern is positivity by spectral overlap, you will be better served by using a fluorescence-minus-one control (FMO), in which all antibodies are included except the one you suspect is most prone to error from spectral overlap.
4. You should absolutely not be using them to determine positive versus negative cells, or as a gating control in your assay.
5. Keep in mind that the best way to avoid high levels of background staining of antigen-negative populations is to carefully titrate your reagents to ensure the highest positive signals in bright populations, while reducing spread in the negative population.
6. If you are using your isotype control and you are seeing high levels of non-desired staining, it is time to look carefully at your blocking step in your protocol. Are you using an Fc-block if you have myeloid cells? Have you tried adding excess immunoglobulins or whole serum to your buffer? Each of these can help pull down your nonspecific adherence.
7. Are you certain it is non-specific antibody adherence you are are dealing with and not free fluorochrome adherence? You can find out by using an isoclonic control. If you add massive amounts of non-fluorochrome conjugated monoclonal antibody to your staining reaction, your fluorescence should drop. If it does not, your issue is not due to nonspecific antibody binding, but to free fluorochrome binding.
8. For cell signalling and cytokine staining, besides the FMO control, make sure not to neglect a biological negative control, whether that be unstimulated cells, or cells treated with an inhibitor of phosphorylation.
9. Don’t forget to use a viability dye in the polychromatic panel. With the proliferation of these dyes in different colors and for both viable and fixed cells, there is no reason to not use these dyes. Viability dyes are essential for removing dead cells that will non-specifically uptake antibodies.
In conclusion, isotype controls are useful for demonstrating that there was poor blocking of the cells. They should never be used to determine or set positivity in fully stained samples.
To learn more about which controls you should use for your flow cytometry experiments and to get access to all of our advanced materials including 20 training videos, presentations, workbooks, and private group membership, get on the Flow Cytometry Mastery Class wait list.

ABOUT TIM BUSHNELL, PHD
Tim Bushnell holds a PhD in Biology from the Rensselaer Polytechnic Institute. He is a co-founder of—and didactic mind behind—ExCyte, the world’s leading flow cytometry training company, which organization boasts a veritable library of in-the-lab resources on sequencing, microscopy, and related topics in the life sciences.
More Written by Tim Bushnell, PhD











