How To Detect Microvesicles, Microparticles, And Ectosomes By Flow Cytometry

There seems to be a big fuss about small flow these days.
What is small flow?
Microvescles, microparticles, ectosomes—the terminology is all over the place but one thing is certain…
There is a need for flow cytometric analysis of extracellular vesicles.
To avoid ambiguity in this post, the population of extracellular vesicles discussed here shall be called microvesicles.
These small, membrane bound fractions of cells released during activation and apoptosis are in the 0.1 to 1 micron diameter size range (or there about depending on which article you read).
Released in vast quantities and carrying a variety of traits from the originating cell these microvesicles are known to be involved in a wide range of processed from stem cell renewal and tumor metastases through to coagulation and inflammation, just to name a few!
They key is that microvesicles have a huge potential to be important biomarkers in early disease detection and/or treatment progression.
What’s Down There? Debris Or Microvesicles?
Microvesicles have been known about for a long time, being dismissed by the regular flow cytometrist as debris or platelet dust and quickly gated out of any subsequent analysis.
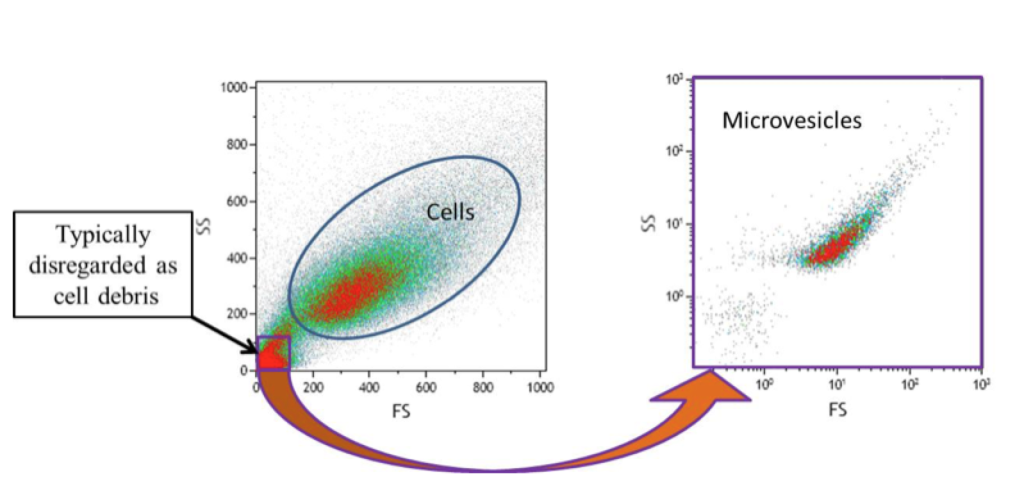
There is increasing interest in seeing what is down there. As in, what is down in the lower left-hand corner of the above figure.
Is it debris? Is it platelet dust? Is it a population of microvesicles?
Or is it all three?
The above figure is a representation of what we are missing when we ignore events in that range.
It’s important to note that the sample on the right in the above figure was further processed before re-analysis in the microvesicle range.
4 Microvesicle Flow Cytometry Mistakes To Avoid
Microvesicles originate from cells and have the same analysis requirements as cells.
For these and other reasons, flow cytometry is a popular choice for microvesicle analysis. However, there are pitfalls with small particle flow cytometry that have led to many conflicting publications.
The only way to avoid these mistakes is to first identify them and then take measures to prevent them.
The following are 4 common mistakes researchers make when preparing microvesicle flow cytometry experiments, as well as how to prevent these mistakes…
1. Not accounting for flow cytometry protocol discrepancies.
Before getting started with your microvesicle experiment, you must take into account pre-analytical variations. When the particles are this small, even the slightest protocol variations can affect your experiment.
Centrifugation speed and duration, collection technique, storage, staining protocols (in particular antibody panels and titration levels) are just a few of the things you should consider.
Minor differences in these protocol steps can lead to huge discrepancies in downstream analysis.
2. Using beads to define microvesicle size ranges.
When setting up for the analysis of microvesicles, many people choose flow cytometry beads to define a size range.
The problem here is, in terms of standards, the different refractive index between beads and microvesicles becomes troublesome.
For example, a polystyrene bead may have a refractive index of about 1.59 whereas while the refractive index of many microvesilces are around 1.39 (for reference, water is 1.33).
Beads scatter a lot more light compared to microvesicles of the same diameter, reportedly up to 100 times!
Here’s the lesson—just because you can see a 0.2 micron diameter bead by flow does not mean you can see a 0.2 micron diameter microvesicle by flow.
3. Failing to acknowledge the possibility of unseen information.
Microvesicles are between 0.1 – 1 micron diameters in size. Though most fall in the smaller end of that range.
Commercial cytometers can struggle to detect light from a 0.2 micron diameter bead. As discussed in point #2 above, beads scatter a LOT more light than microvesicles.
With this in mind you have to ask yourself, how many microvesicles can I actually see by flow cytometry?
According to many flow cytometrists—not a lot.
When analyzing microvesicles by conventional flow cytometry, it’s worth considering the massive amount of information that goes unseen.
Don’t assume you’re seeing all the microvesicles in your experiment. Instead, constantly ask yourself, “What am I NOT seeing?”
As techniques improve, you’ll be able to see more and more information and continue to get a clearer picture of what’s really going on in your microvesicle flow cytometry experiments.
4. Failing to account for coincident events.
Accounting for coincidence events is important to every flow cytometry experiment, including experiments measuring and reporting microvesicles.
This is especially true if you’re hoping to publish the flow cytometry data you obtain.
The problem here is that conventional doublet discrimination in this microvesicle size ranges is exceedingly difficult.
A single event, or dot, on a flow cytometry plot may actually be many, many microvesicles passing through the laser beam at the same time. The only way to know for sure is to check.
One way to check your microvesicle analysis is with serial dilutions.
If you’re able to see a stable fluorescent signal with dilutions resulting in a lower event count, you can gain more certainty over the assumption that you’re analyzing (mostly) single events.
The analysis of microvesicles by flow cytometry is not easy. However, until better instrumentation is developed, in wide use, and shown to be more accurate, using conventional flow cytometry to detect and measure microvesicles is of great value. While a multi-platform strategy continues to be the wisest approach, much information on microvesicles can be gained by conventional flow cytometry.
To learn more about detecting microvesicles by flow cytometry and to get access to all of our advanced materials including 20 training videos, presentations, workbooks, and private group membership, get on the Flow Cytometry Mastery Class wait list.

ABOUT TIM BUSHNELL, PHD
Tim Bushnell holds a PhD in Biology from the Rensselaer Polytechnic Institute. He is a co-founder of—and didactic mind behind—ExCyte, the world’s leading flow cytometry training company, which organization boasts a veritable library of in-the-lab resources on sequencing, microscopy, and related topics in the life sciences.
More Written by Tim Bushnell, PhD











